Spectral Library Search

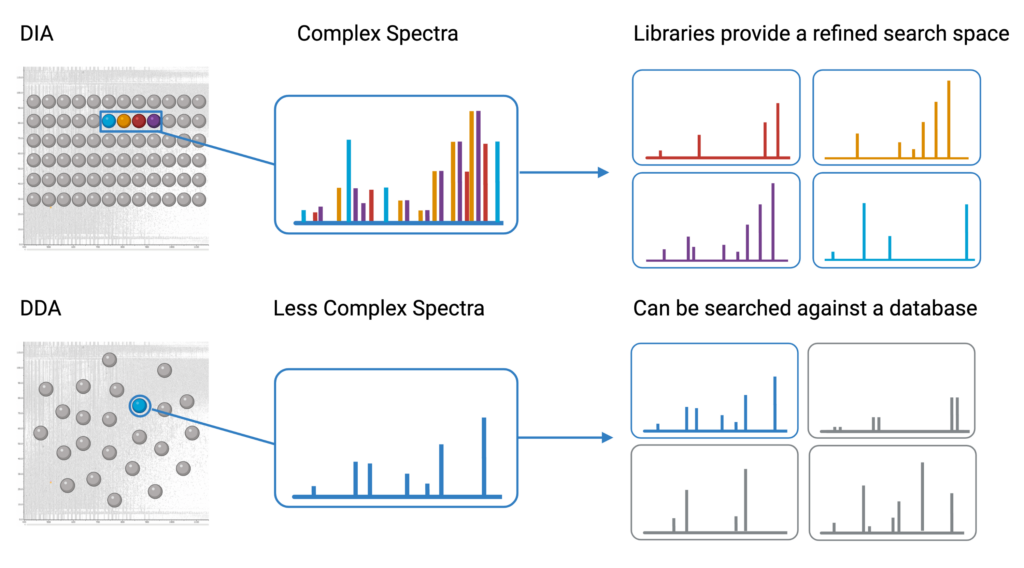
PEAKS 提供 基于最新深度学习技术的先进谱库搜索功能,可从复杂的 数据非依赖分析(DIA)谱图 中鉴定肽段 ¹。由于 DIA 谱图包含 前体质量窗口内的离子信息,因此会同时收录多种肽段的碎片离子。这使得 DIA 谱图的复杂程度远高于 数据依赖分析(DDA)谱图。
为了从这类复杂谱图中准确鉴定肽段,谱图库 提供了一个 精炼的搜索空间:它将实验数据的 碎片离子模式、预期保留时间及其他区分特征,与库中已鉴定的谱图进行匹配。
借助 PEAKS 一体化软件,用户可在 同一平台完成全流程分析(从谱图库构建到谱库搜索),无需在 “项目结果转换” 和 “评分系统” 之间牺牲时间或精度。
灵敏准确的谱图库构建
谱图库搜索流程的第一步,是利用 数据依赖采集(DDA)质谱数据集 构建谱图库。
建议采用 分馏样品 构建谱图库,以实现研究物种的 最深蛋白质组覆盖度 ,并获得最多的肽段数。谱图库一旦建成,可用于 多次数据非依赖分析(DIA) ;因此,投入物料和仪器时间来构建一个全面的谱图库,往往是值得的。
随后,将该 DDA 数据导入 PEAKS 软件,利用 精准且灵敏的 PEAKS DB 蛋白质数据库搜索算法 进行检索。可通过 靶标 – 诱饵数据库搜索估算的假发现率(FDR) ,控制鉴定肽段的置信度阈值。随后,利用 高置信度鉴定的肽段 ,即可构建谱图库。
谱图库中的 每个肽段条目 ,都会承载来自 DDA 谱图的 关键信息 —— 这些信息为在复杂 DIA 质谱样品中鉴定该肽段,提供了最佳识别依据。
从全谱图中,会提取以下数据: 前体质量 、 离子淌度(若仪器支持) ,以及与该肽段匹配的 碎片离子强度和质荷比(m/z) 。此外,谱图库中每个条目的 保留时间(RT) 会被转换为 索引保留时间(iRT) :iRT 通过软件 内置肽段库 生成,因此在构建谱图库和开展 DIA 分析时, 无需额外添加 iRT 标记肽 。
若质谱仪具备 离子淌度分离肽段 的功能,相关信息也会被纳入谱图库(需启用 PEAKS IMS 附加模块 ),进一步提升谱库搜索的灵敏度。
如何生成谱图库
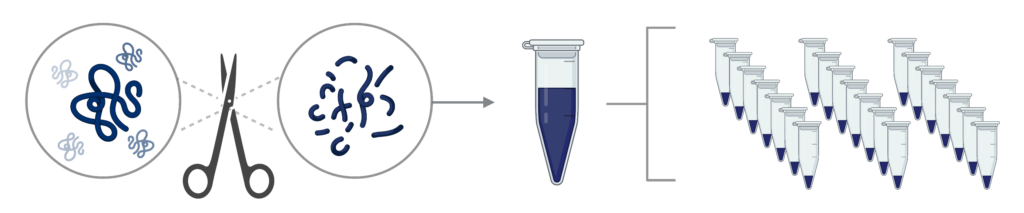
Step 1 – Sample preparation: 蛋白质被酶解为肽段,随后这些肽段可进行分馏。
Step 2 – LC-MS/MS: 肽段通过数据依赖分析(DDA)进行分析,以生成谱图。
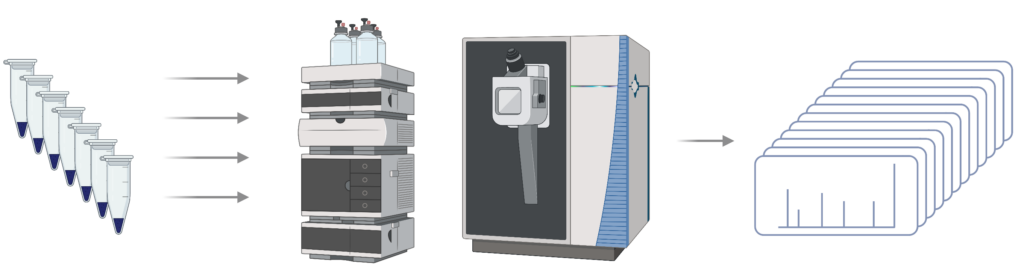
Step 3 – Library generation: DDA 数据通过 PEAKS DB 进行分析,并导出为谱图库,用于后续的 DIA 数据分析。

借助由深度学习驱动的保留时间预测算法,最大化谱库匹配率
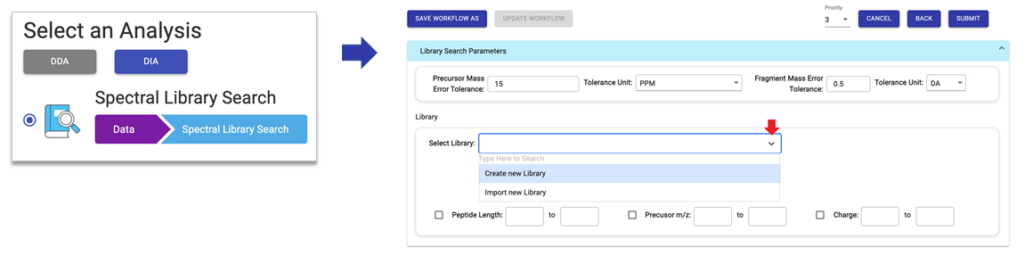
谱库搜索本身使用起来非常简单。谱图库既可以在创建新项目前预先设置,也可以在谱库搜索参数设置过程中进行设置。
准备执行搜索时,只需将目标数据添加到项目中,选择谱库搜索工作流程,然后指定所需的谱图库并设置相应的搜索参数即可。
对于大规模蛋白质组学研究项目,推荐使用 PEAKS Online—— 它不仅能处理更大规模的样本队列,还能为这些全面的 PEAKS 谱库搜索提供一个高通量、可扩展的平台。
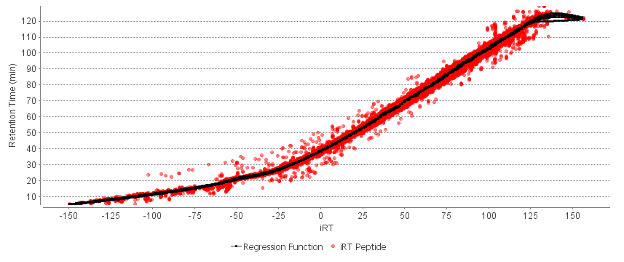
PEAKS 采用基于机器学习开发的先进肽段特征检测算法,利用液质联用(LC-MS)信息预测样品中肽段的前体质量和洗脱曲线。
结合谱图库中的详细信息,以及基于深度学习开发的保留时间预测算法,可对所搜索的 DIA 数据集中谱图库肽段的洗脱时间进行预测。
谱图库中的肽段会与数据集中的肽段特征进行匹配,而匹配质量的评分则基于谱图库谱图与所搜索的相应二级质谱(MS/MS)谱图之间的拟合程度。
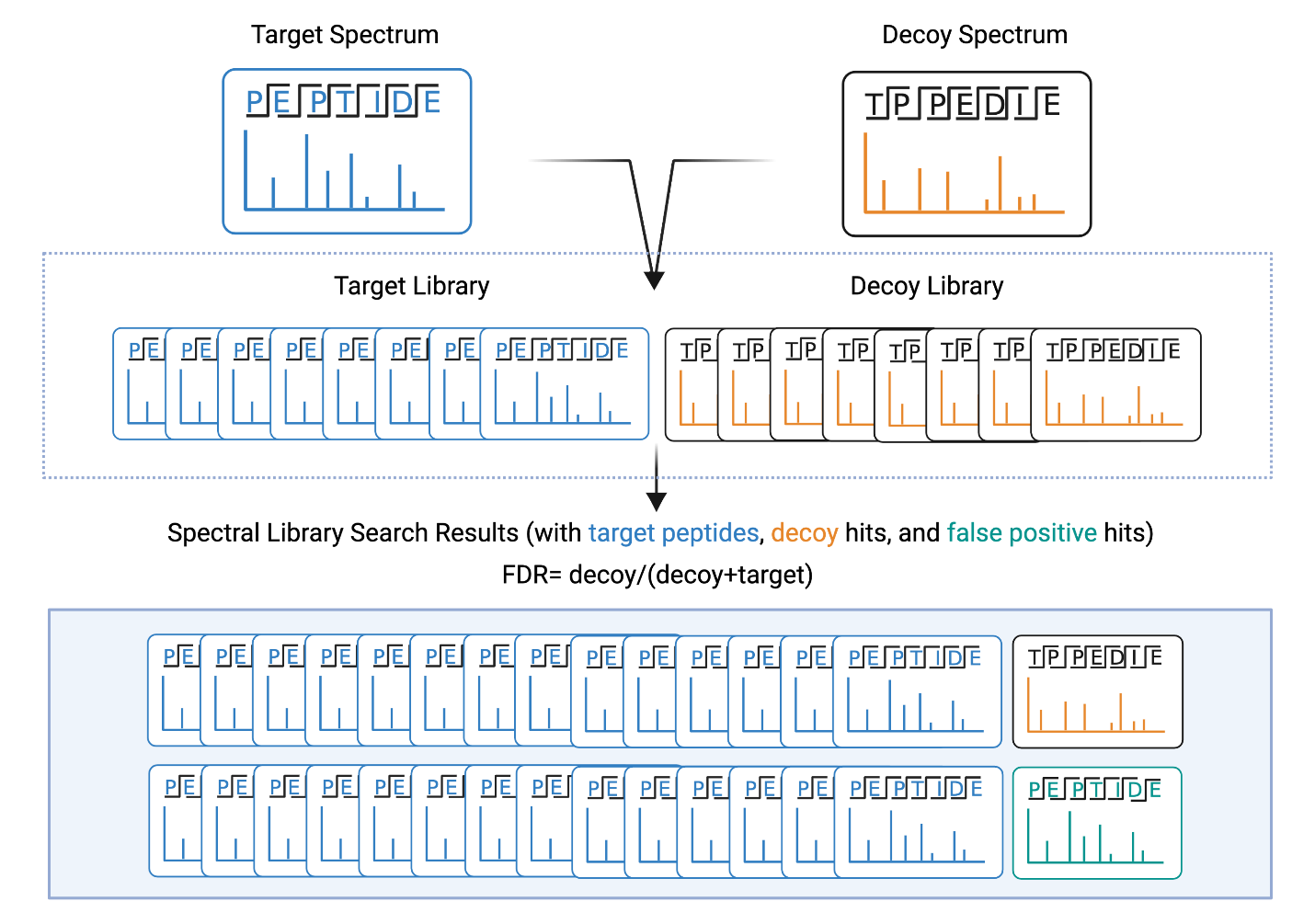
为确保结果的准确性,假发现率(FDR)通过诱饵谱图搜索进行预测(图 2)。
利用谱图库中的肽段,软件会生成打乱的肽段序列,同时将其相关的碎片离子列表也进行打乱,以构建诱饵谱图。因此,当执行谱库搜索时,软件所搜索的谱图库中,靶标谱图和诱饵谱图的数量是相等的。
基于结果中匹配到的谱图,可估算假发现率,并设置置信度阈值。关于 FDR 的更多细节,请参见我们的FDR tutorial.
结果是一份从 DIA 谱图中精确匹配到对应库谱图的肽段列表,作为鉴定依据。通过 FASTA 文件可推断出蛋白质,为鉴定结果提供生物学背景。
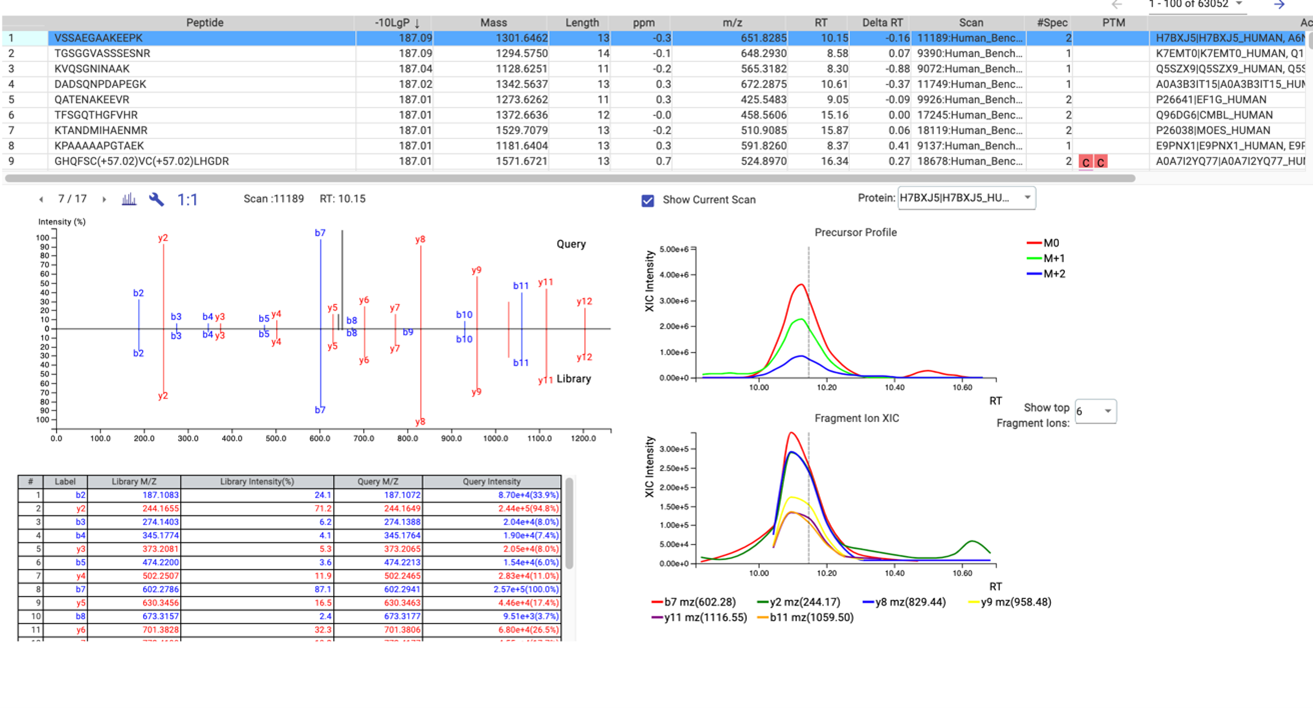
借助 PEAKS 易用的界面,谱库结果以图形化方式呈现(便于直观解读),并配有详尽的表格(供深入分析)。这些谱库结果可用于后续的定量分析。结果视图示例如下(图 3)。
参考文献
- Tran NH, Qiao R, Xin L, Chen X, Liu C, Zhang X, Shan B, Ghodsi A, Li M. Deep learning enables de novo peptide sequencing from data-independent-acquisition mass spectrometry. Nat Methods. 2019 Jan;16(1):63-66. doi: 10.1038/s41592-018-0260-3. Epub 2018 Dec 20. PMID: 30573815.