

自下而上的蛋白质组学完整解决方案
PEAKS® Studio 提供完整的自下而上的蛋白质组学解决方案,具有更高的准确性、灵敏度和速度。能够全面支持数据依赖采集(DDA)和数据非依赖采集(DIA)数据,并针对多种应用场景更新了相应的工作流(如经典和非经典多肽/蛋白质的深度鉴定),为用户提供全面的解决方案,助力研究迈向新高度!
One-Stop
Discovery and Targeted Proteomics
Comprehensive
and Confident
DDA Solutions

High Accuracy and Sensitivity DIA Solutions
High Precision
PRM Solutions
de novo and
DeepNovo
Algorithms
Vendor Neutral
Software
联系我们获取PEAKS Studio 13.1 !
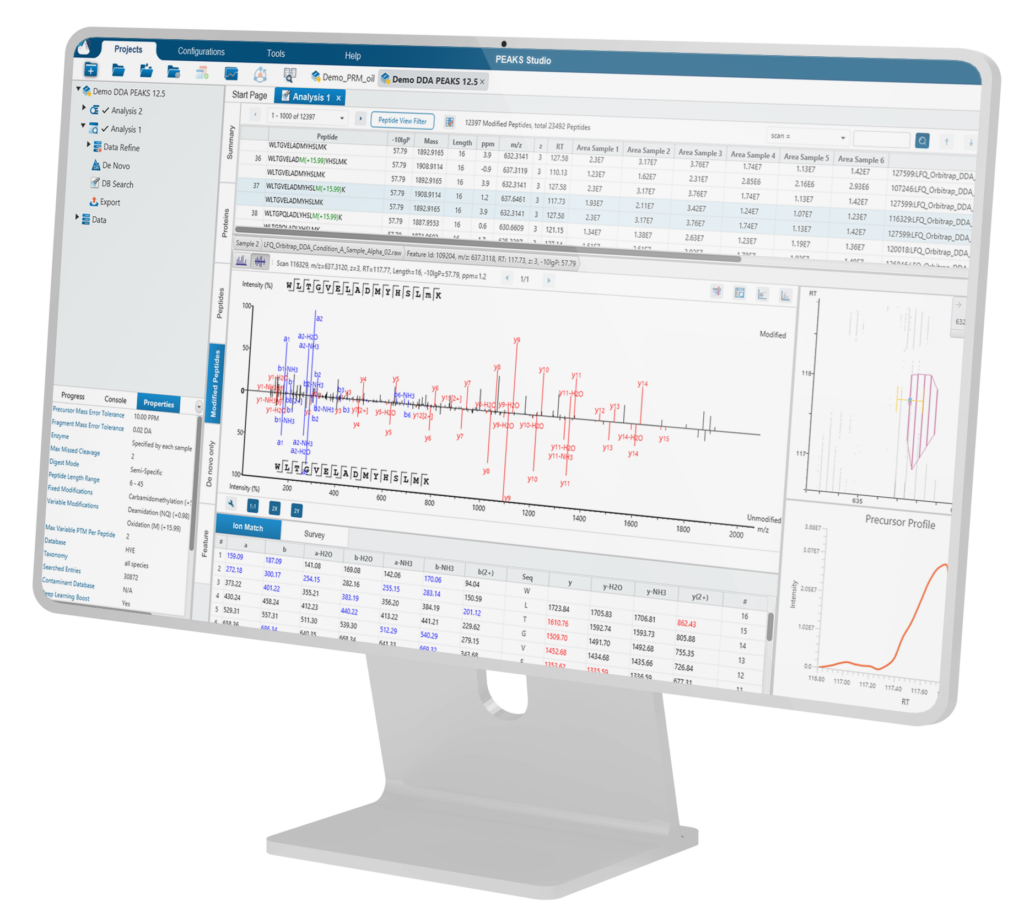
全新设计的DDA和DIA蛋白质组学和多肽组学工作流
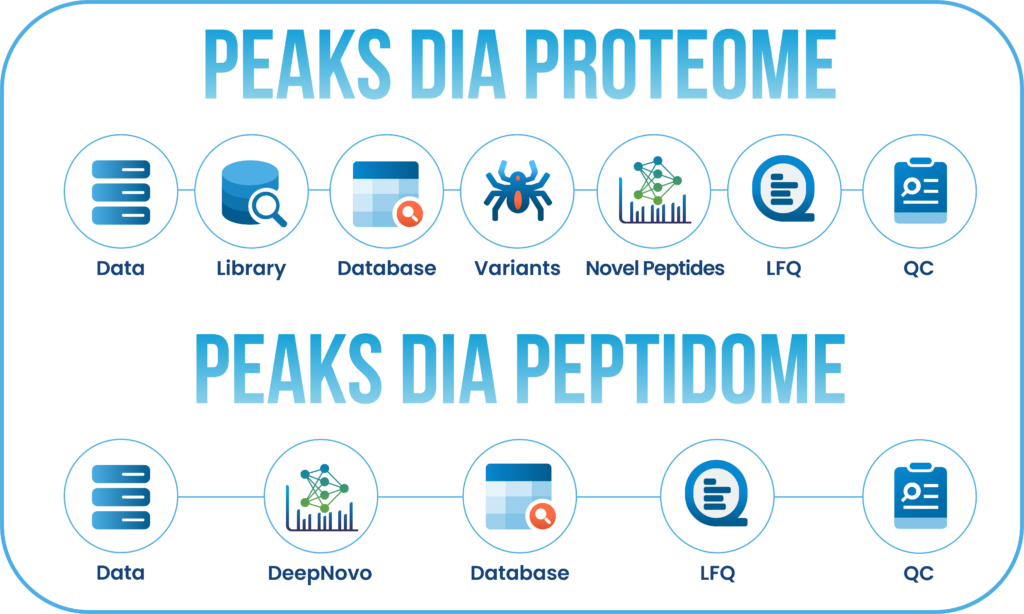
PEAKS Studio 的DDA和DIA工作流已精简为两大强大选项:蛋白质组学(Proteome)和肽组学(Peptidome)。
Proteome工作流能够支持传统的蛋白质定性和定量分析,结果聚焦于蛋白质水平。而肽组学工作流利用基于DeepNovo深度学习算法的从头测序技术,结合多肽数据库搜索与序列变体分析,实现肽段水平的深度挖掘分析。
用户可自定义分析流,可根据分析需求在任意步骤选择提交分析,并可在此分析基础上添加或修改其他步骤。分析结果将以直观的结果节点形式展示,便于用户解读、探索和验证数据。

核心功能
DIA Workflow

LC/MS Data
Refine View

Feature-Based Identification
DeepNovo
Peptidome
Confident
PTM/Mutations
Quality Control (QC) Function
DIA Feature
View
Quantification
View

Hybrid DIA
and PRM
Protein &
Peptide View
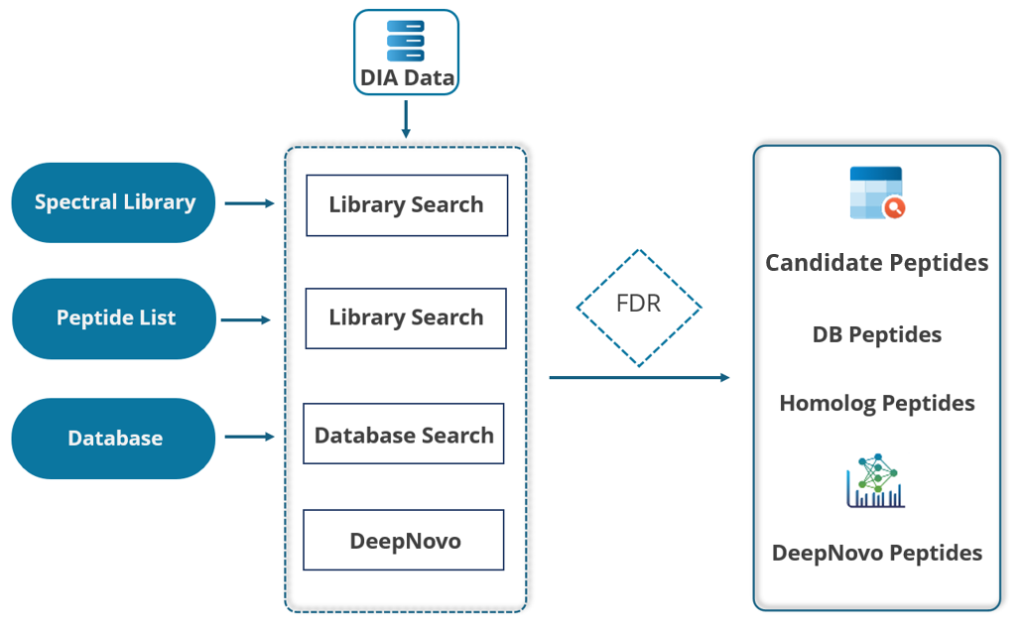
整合直接序列数据库搜索的一体化DIA工作流
PEAKS® Studio 提供独特的DIA工作流,通过整合四种方法最大限度地提升肽段鉴定率:谱图库搜索、直接数据库搜索和从头测序及序列变异分析,定性定量提升高达 20%,同时优化 CPU 和 GPU 资源的数据处理速度。
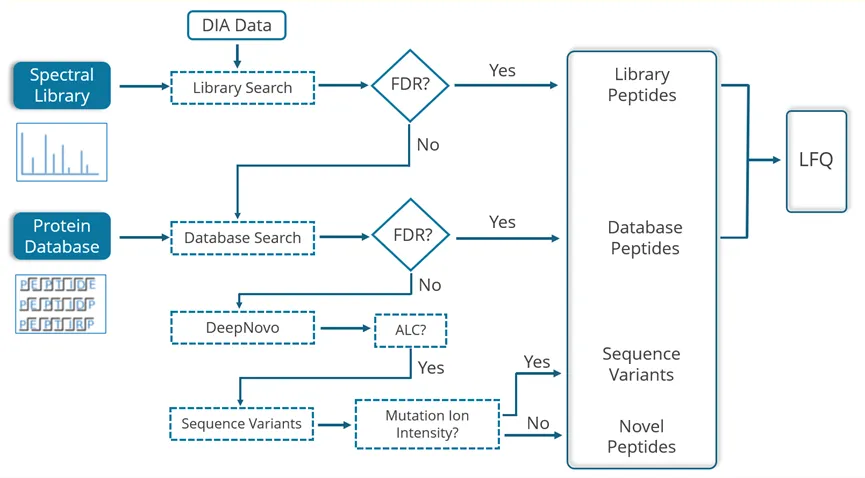
- SL search是针对于预定义的谱图库进行的。不包含在谱图库中的多肽可以用direct DB的方法直接对蛋白的序列数据库继续搜索。
- DIA数据可直接搜索蛋白质序列数据库。先进的机器学习算法显著提升了肽段鉴定的准确性和灵敏度。PEAKS DIA工作流现已支持用户指定的任意翻译后修饰(PTM)鉴定。这一功能将大幅提升修饰肽段的鉴定数量,且无需依赖谱图库中预先存在的修饰肽段条目。
- 在序列数据库中未匹配的谱图,将进行从头测序。
- 利用 SPIDER 算法将高分从头测序肽段与数据库比对,以发现序列变体。
- 无论是来自于谱图库搜索还是直接序列数据库搜索的多肽鉴定结果,都可以进入到下一步的定量分析。
谱图库搜索和直接序列数据库搜索均是可选的。用户可单独执行谱图库搜索、直接序列数据库搜索,或按顺序组合执行这两个搜索分析。
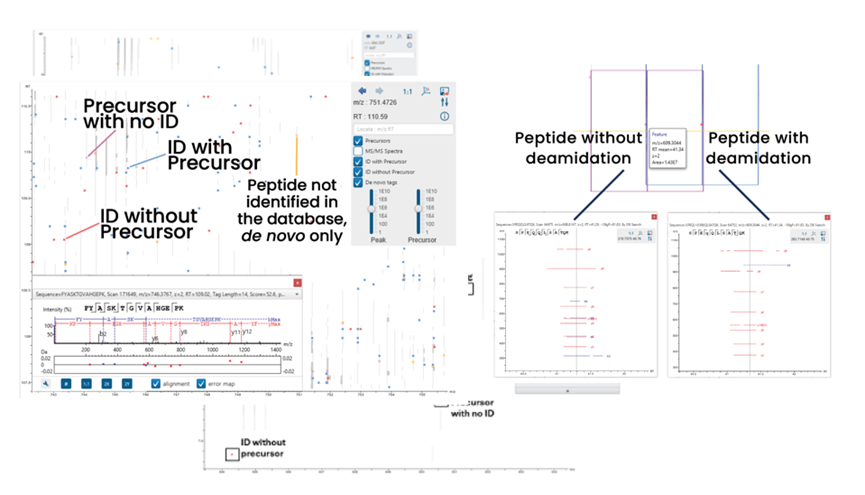
LC/MS Data Refine视图
用户可在LC/MS谱图上轻松可视化展示母离子、有/无关联母离子的鉴定结果以及从头测序标签,从而显著提升结果验证的准确性和效率。
对于一个给定的肽段,可以在多肽列表和LC/MS视图之间无缝切换。点击一个或多个鉴定结果即可显示并比较支持的碎片离子,这便于验证重叠的肽段鉴定。
支持的碎片离子还会在肽段选项卡的LC/MS谱图中显示。用户也可点击完整 LC/MS 谱图上显示鉴定结果。
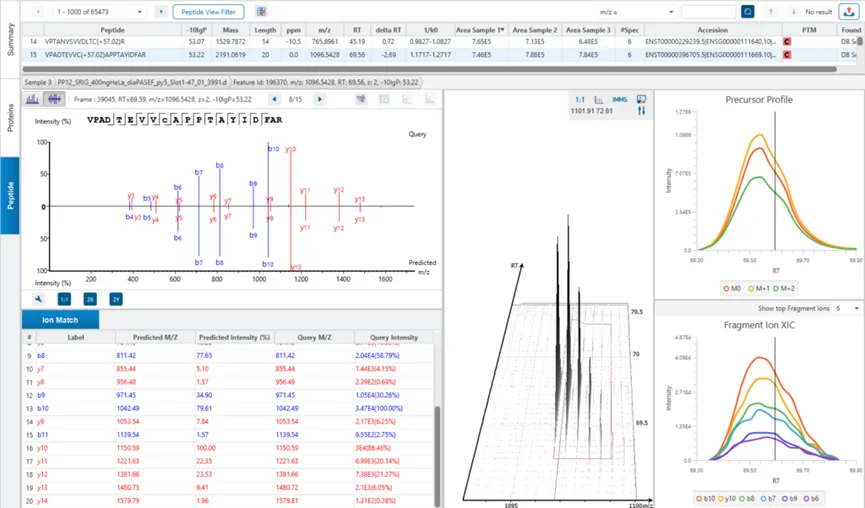
基于特征峰的定性分析
- 基于MS1特征峰的鉴定可提高灵敏度和肽鉴定效率。
- 专为DDA设计,提高了数据分析的重现性。
- 整合了数据库搜索和从头测序,实现深度蛋白质组学。
- PEAKS DDA的启用深度学习增强功能,使多肽的鉴定达到最大化。
- 了解更多关于de novo测序为您的研究带来的优势。
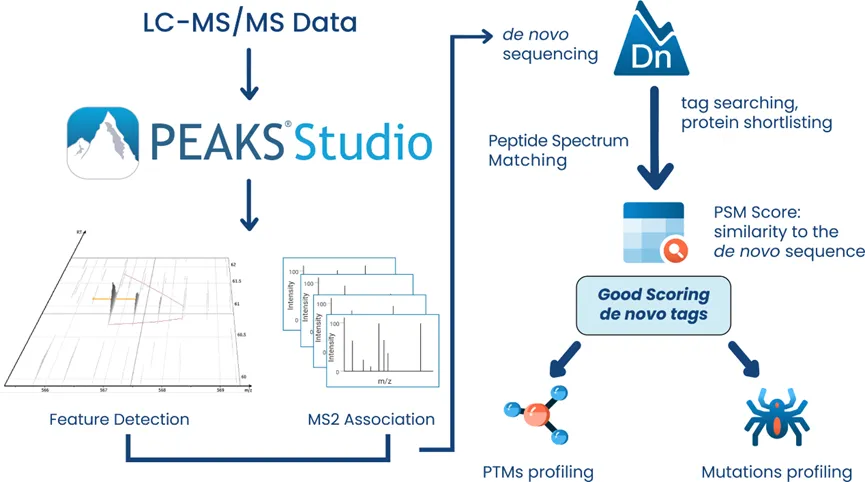
PEAKS DeepNovo多肽组学:先进的免疫肽组学解决方案
该工作流是专门为多肽组学研究设计的,结合了数据库搜索、从头测序和多肽突变的鉴定。DeepNovo肽组学工作流旨在通过整合全面的基因数据库,弥合蛋白质组学与基因组学之间的鸿沟。该DDA工作流支持非经典参考序列,能够识别标准经典参考数据库中常被遗漏的变异肽段和修饰肽段。DIA肽组学工作流现已支持谱图库和目标肽段列表功能,可专门针对样本中预期存在的目标肽段进行精准检索。
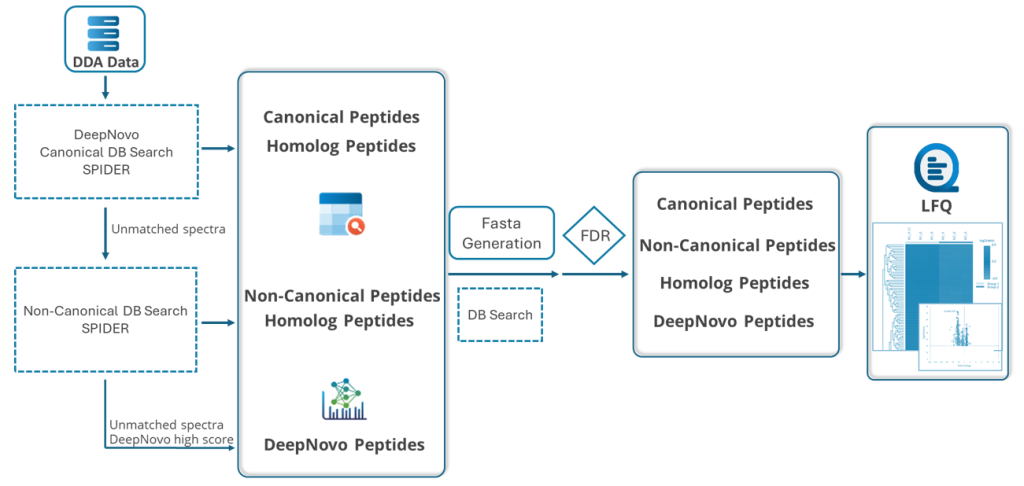
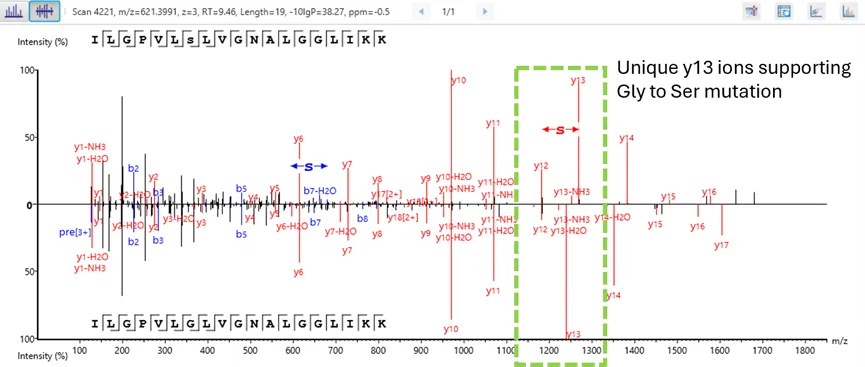
更高水平的置信度:精确到氨基酸的可信翻译后修饰/突变位点鉴定
翻译后修饰(PTM)与突变分析工作流程通过基于氨基酸水平的实验证据,实现了对PTM位点和突变位点的高精度鉴定,从而提供更高置信度的分析结果。
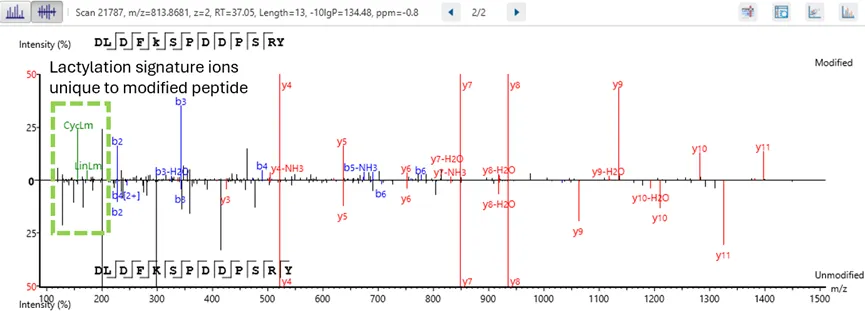
这一功能使研究人员能够精确定位蛋白质内部的特定修饰位点和突变位点,从而对蛋白质功能与调控机制获得细致而准确的理解。通过运用强大的算法和全面的数据分析策略,该工作流可以确保翻译后修饰和突变位点的鉴定具有极高的置信度,有助于深入探究蛋白质动态变化规律和疾病发生机制。这种分析能力对于深化我们对生物学过程的认识以及开发靶向治疗策略至关重要。
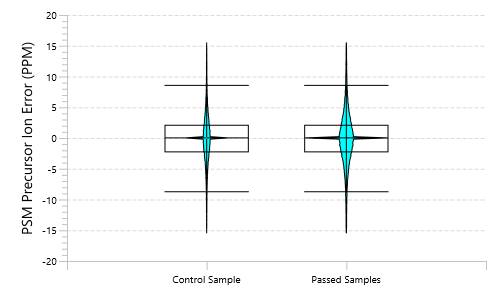
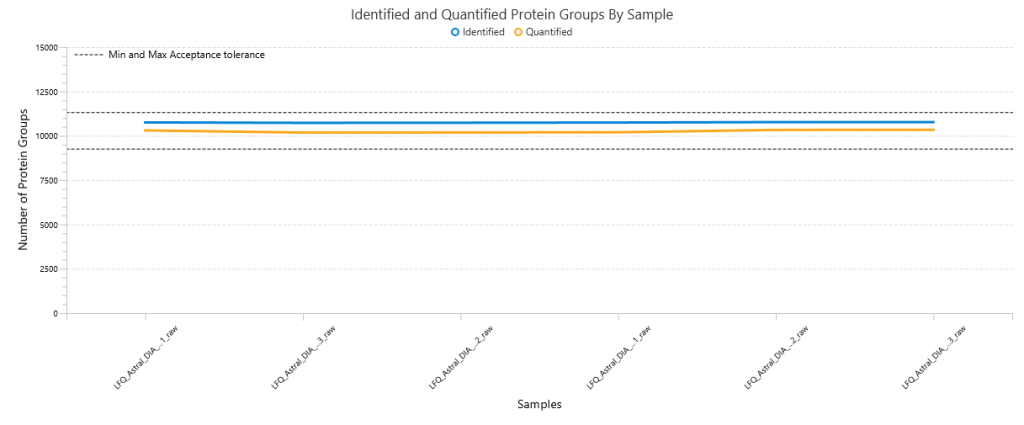
质量控制(QC)功能:从原始数据到结果的深入分析
在PEAKS® Studio中,质量控制(QC)分析功能使用户能够全面评估原始数据和/或分析结果的统计信息,深入洞察LC-MS采集过程的各项属性特征。这一自动化工具同时适用于DDA和DIA模式,可提供关键参数用于判定数据质量并评估实验方案设置的合理性。
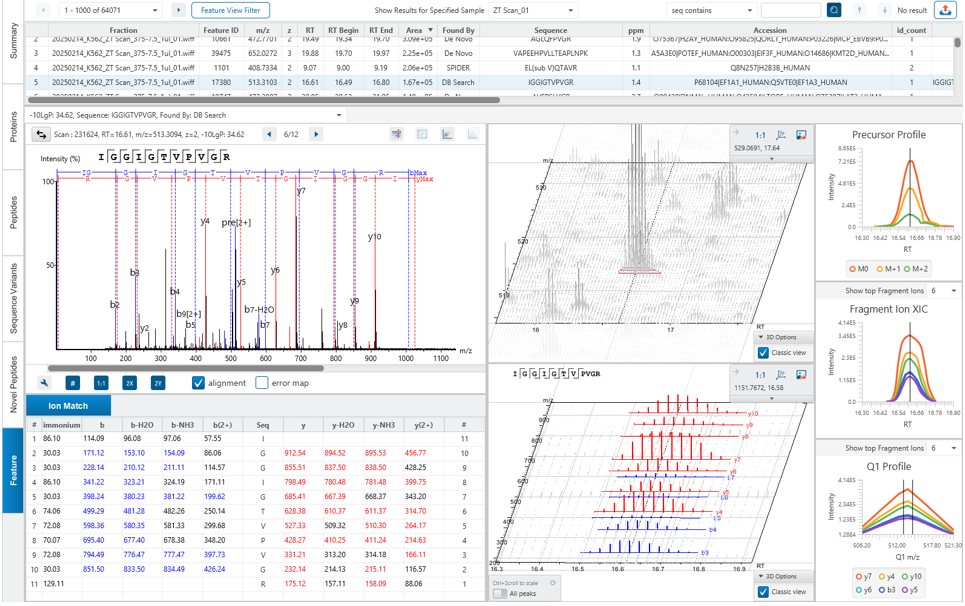
DIA Feature View
DIA特征峰视图体现了PEAKS致力于提供完整结果透明度的设计理念,使用户能够深入调查、验证和确认前体离子。该视图完整展示所有特征峰(包括已鉴定和未鉴定的),提供所有碎片离子归属信息、镜像图谱(用于展示实验观测碎片与预测碎片之间的相关性),以及MS1和MS2特征视图。
Quantification View
通过PEAKS Q附加模块,PEAKS Studio还能够在无需预先了解相关蛋白质的情况下,同时测定一组样本中蛋白质相对丰度的变化。在PEAKS中,通过PEAKS PTM或SPIDER算法搜索发现的肽段将被纳入定量分析步骤。
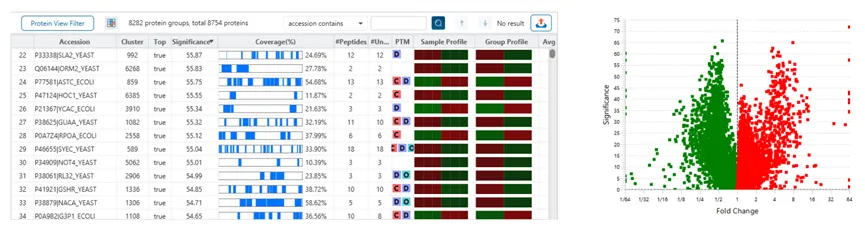
如果选择了“Feature-based”的定量,LFQ结果将包含一个新的“de novo only”选项卡,其中包含与数据库不匹配的肽段的定量结果。
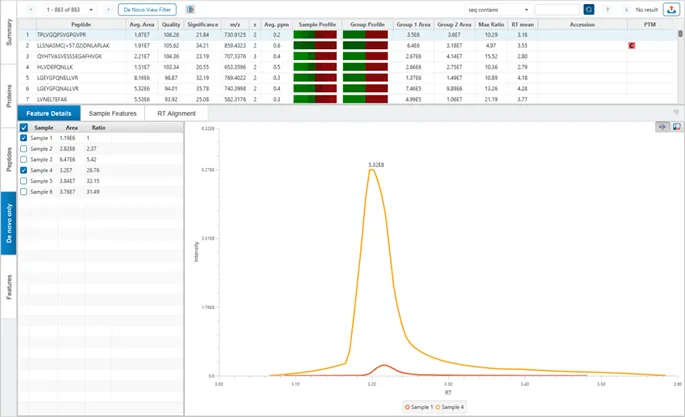
Hybrid DIA
Thermo的Hybrid-DIA技术,仅在PEAKS® Studio提供,结合了靶向和发现驱动的方法,使其成为临床蛋白质组学应用的理想选择。
Hybrid-PRM/DIA是一种新的数据采集策略,通过智能触发多并行反应监测(PRM),结合使用实现DIA对临床生物标本进行发现驱动的数字化,可以提高对特定分析集的灵敏度。重标记参考肽被用作PRM和内源性肽监测的触发。
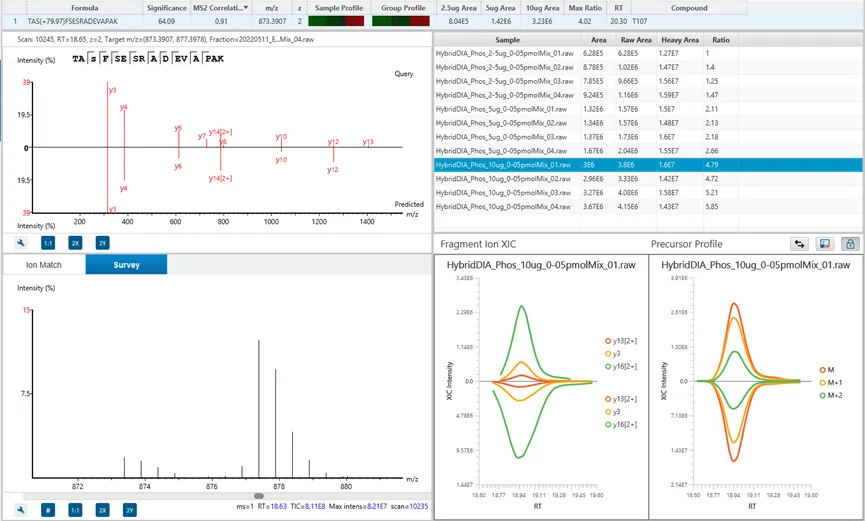
The Protein View
蛋白质视图在DDA和DIA工作流程中展示了复杂生物样品的蛋白谱。对于每个蛋白质,Coverage视图显示了一个包含谱图注释信息的肽图,用于结果的验证。使用PEAKS传统的从头测序辅助数据库搜索的方法,用户可以轻松地在蓝条状图中查看与参考序列数据库匹配一致的多肽序列,而灰色的条状图表示de novo only与参考序列库部分匹配的序列标签。
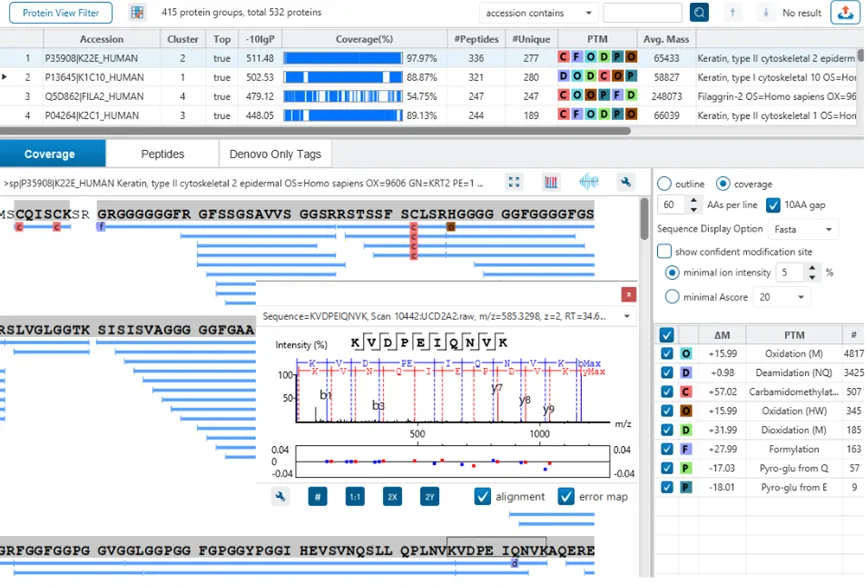
The Peptide View
多肽视图提供了多肽鉴定结果的列表,并且包含了MS1的丰度信息,对每一个修饰肽段,修饰位点的置信度信息( Ascore)也会显示在这里。
无与伦比的客户服务支持
![]() 快速专业的技术响应
快速专业的技术响应
我们内部的专业科学家团队将及时回应您的PEAKS技术咨询,确保您的工作流程和研究进度不受任何阻碍。
![]() 持续更新的教程资源库
持续更新的教程资源库
提供详尽的分步操作指南、流程演示和在线研讨会,帮助您和团队充分发挥PEAKS Studio的分析潜能。
![]() 基于用户反馈的持续优化
基于用户反馈的持续优化
我们认真倾听用户声音。您的建议与需求将直接推动我们后续版本的开发和平台功能改进,确保软件始终满足蛋白质组学研究的前沿需求。

授权信息
PEAKS Studio 可以根据您实验室的使用需求选择授权版本
- Desktop License–24线程, 最多可以处理达到CPU的24线程
- Workstation License–48线程,最多可以处理达到CPU的48线程
系统要求
PEAKS® Studio 13.1建议安装在 Windows 10或更高版本的64 位Windows 操作系统上。
推荐配置
Desktop License: 30+线程处理器、64GB+of RAM以及兼容GPU(如下所述)
Workstation License:60+线程处理器、128GB+RAM以及兼容 GPU(如下所述),如 Intel Core i7/i9/Xeon或AMD Ryzen 7/9/Threadripper处理器
如需运行DeepNovo,需要机器配备NVIDIA CUDA计算能力≥8 且≥8GB内存的GPU。
如需运行DIA Database Search,建议机器配备具有NVIDIA CUDA 计算能力≥5 且≥8GB 专用内存的GPU。
GPU 驱动必须更新到 CUDA 12.3 或更高版本。
References & Resources
References
- Tran NH, Qiao R, Xin L, Chen X, Liu C, Zhang X, Shan B, Ghodsi A, Li M, Deep learning enables de novo peptide sequencing from data-independent-acquisition mass spectrometry, Nat. Methods, 16, 63–66 (2019). https://doi.org/10.1038/s41592-018-0260-3
- Tran NH, Zhang X, Xin L, Shan B, Li M. De novo peptide sequencing by deep learning. Proc. Natl. Acad. Sci. U.S.A. 114, 8247-8252 (2017). https://doi.org/10.1073/pnas.1705691114
Resources
- PEAKS Studio 13 Brochure
- Application Note: The Proteomics Continuum: Discovery, Definition, and Validation-PEAKS solution for translational medicine research
- Application Note: Determination of unknown PTMs and increased protein sequence coverage from high temperature acidic enzyme digests
- Application Note: From Discovery to Targeted Proteomics: A Complete Workflow for MRMHR-based Peptide Quantitation