
PEAKS® Studio 13的DDA和DIA工作流全新升级,流程设置更加简洁,用户体验更加友好,同时分析算法也进一步加强,软件性能大幅提升。PEAKS DIA的分析速度提升数倍,DIA SPIDER新功能遥遥领先,能够发现数据库中未包含的新肽段和序列变异肽,从而实现DIA数据更深度的挖掘。DIA result页面新增的Feature视图,使得DIA结果更加透明,便于交互式谱图校验。
无与伦比的DIA性能
PEAKS全新的高性能DIA解决方案将加速您的发现蛋白质组学研究进展,确保鉴定结果全面而准确,为复杂生物学深入分析提供有力支撑。
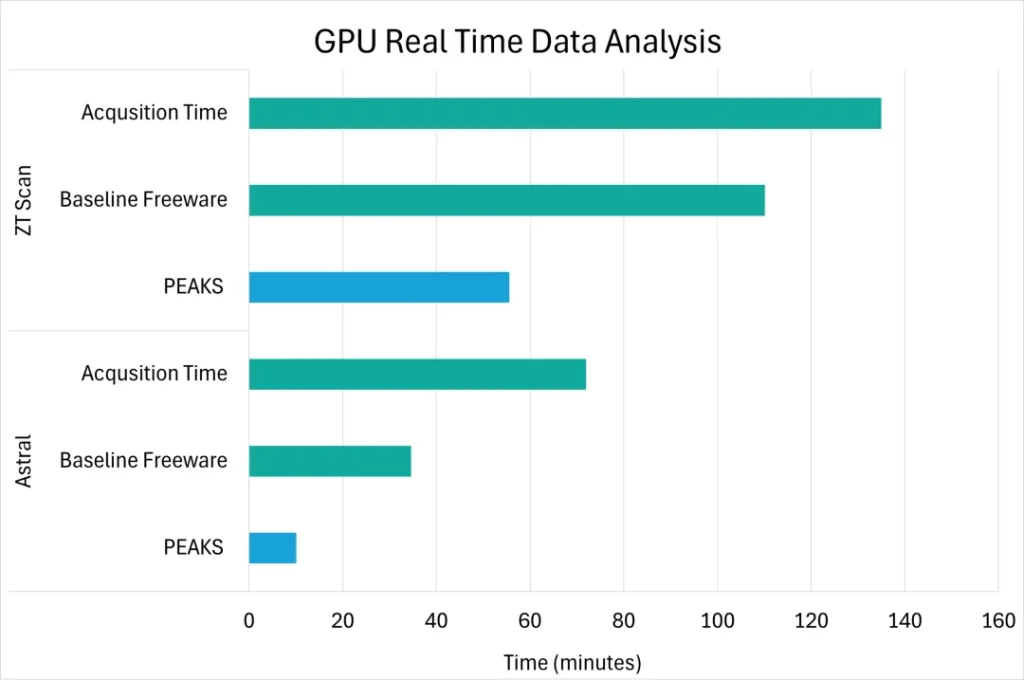
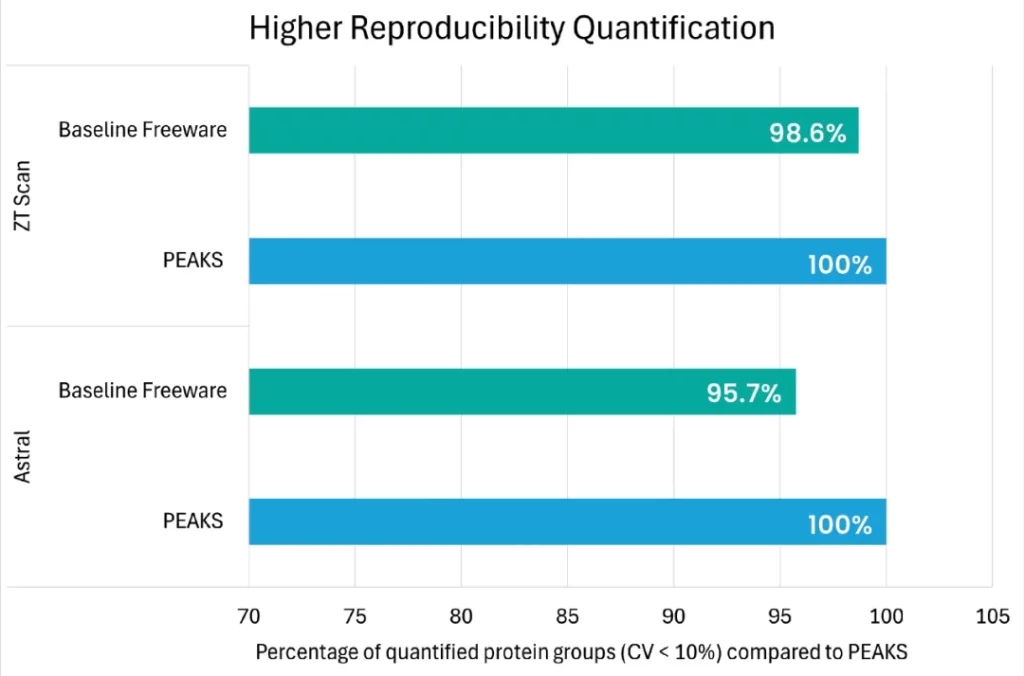
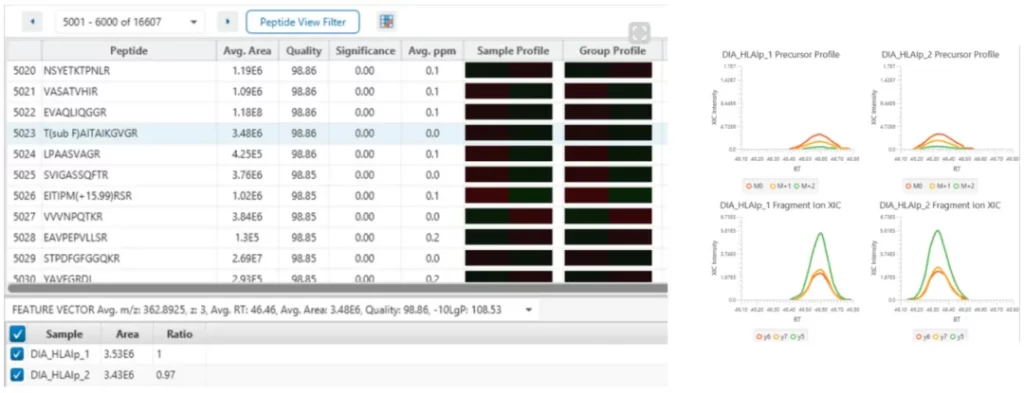
分析结果高度透明
全新“Feature”视图展示了该鉴定肽段相关联的所有MS1特征峰,用户可通过此信息查看、验证和确认结果的可靠性。
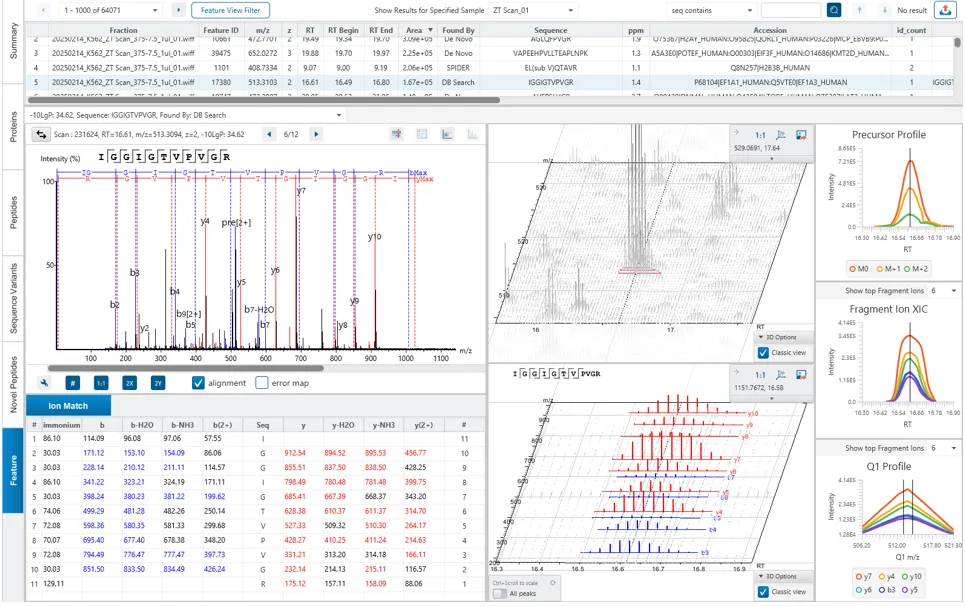
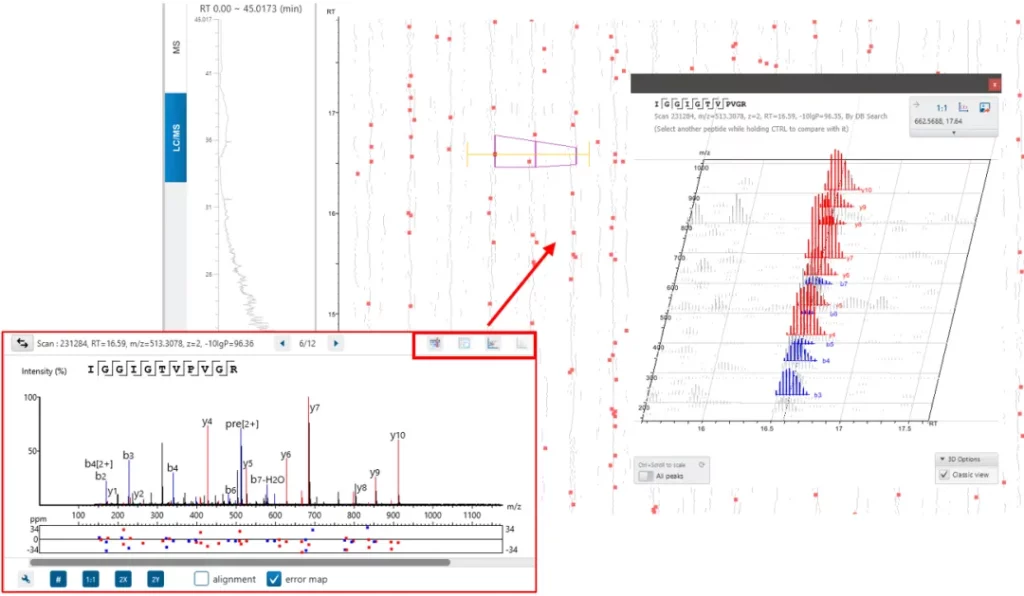
DIA序列变异和新肽发现,解锁蛋白质组学的“暗物质”
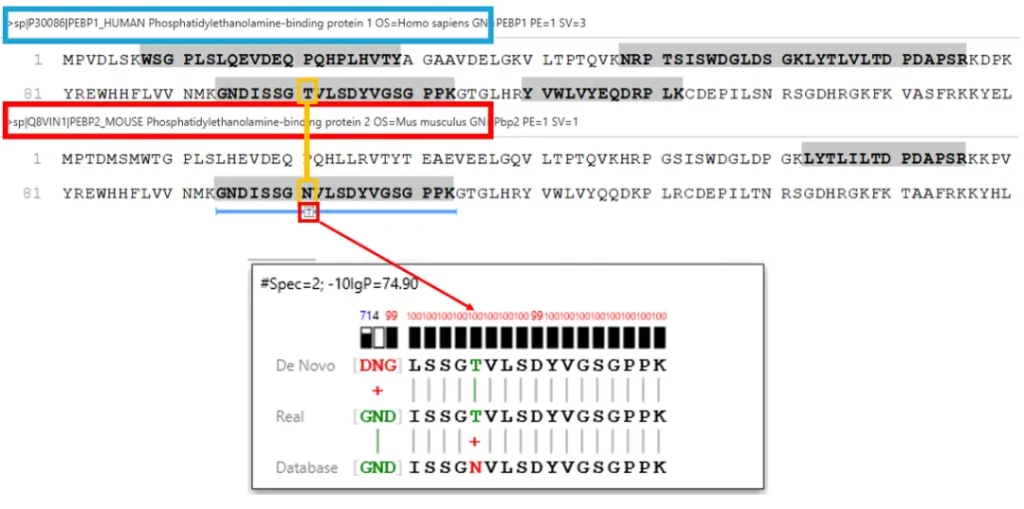
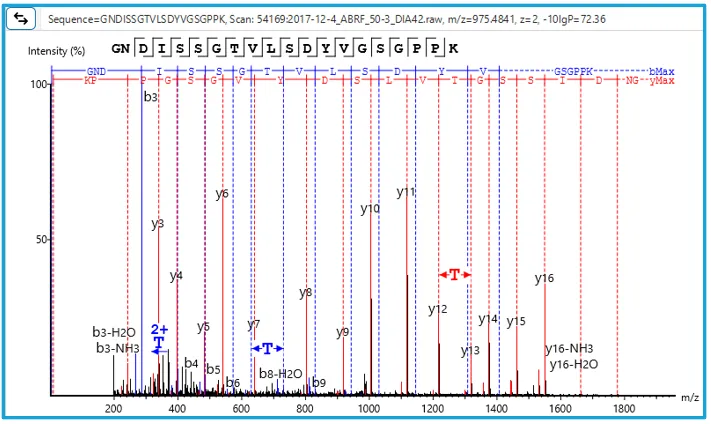
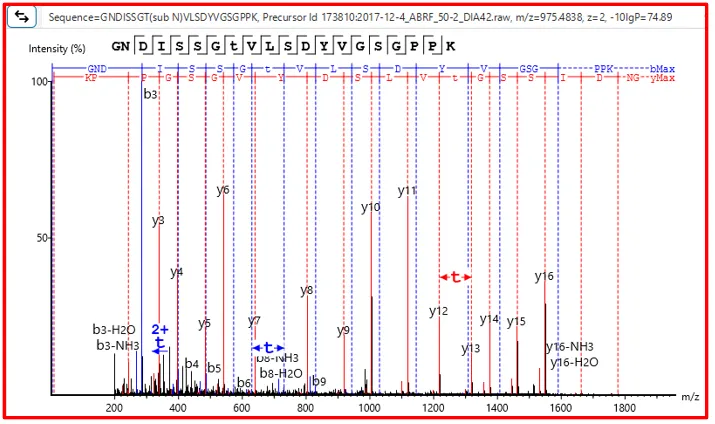
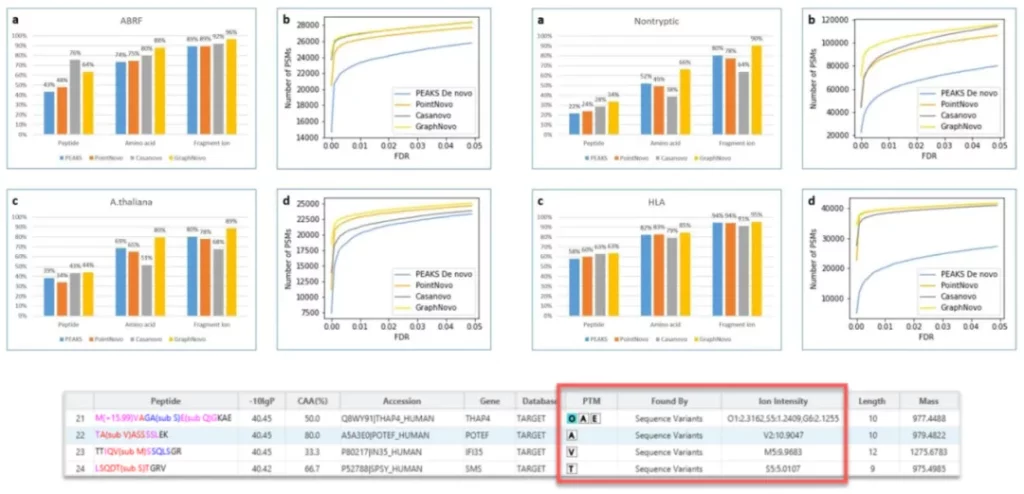
用human和mouse参考数据库同时对人源蛋白的酶切肽段进行搜库分析,结果中human来源的肽段报告为found by DB search,而经过DIA SPIDER分析,与mouse数据库匹配的结果则报告为mutation,表明算法超高的灵敏度。
新功能总览
PEAKS DIA蛋白质组学
>CPU和GPU运算速度均显著提升
>前所未有的高性能,提高灵敏度和准确性
>DIA序列变异分析实现更深度挖掘
>Feature视图提供全面的结果可视化
>优化的蛋白和多肽打分提高鉴定可信度
>蛋白质定量支持Top 3和MaxLFQ方法自由选择
>可自由选择Precursor FDR/score阈值是否应用于结果过滤
PEAKS DIA多肽组学
>应用强大的GraphNovo深度学习测序算法
>DIA Peptidome工作流新增序列变异分析
>LFQ支持数据库匹配和序列变异肽段的共同定量
PEAKS DDA
>全新设计的DDA蛋白质组学和多肽组学工作流,提升使用便利性和灵活性
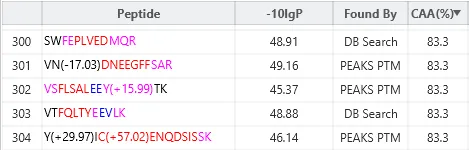
>PEAKS PTM和SPIDER鉴定结果可用于定量分析
>多肽列表中新增“Found by”筛选功能
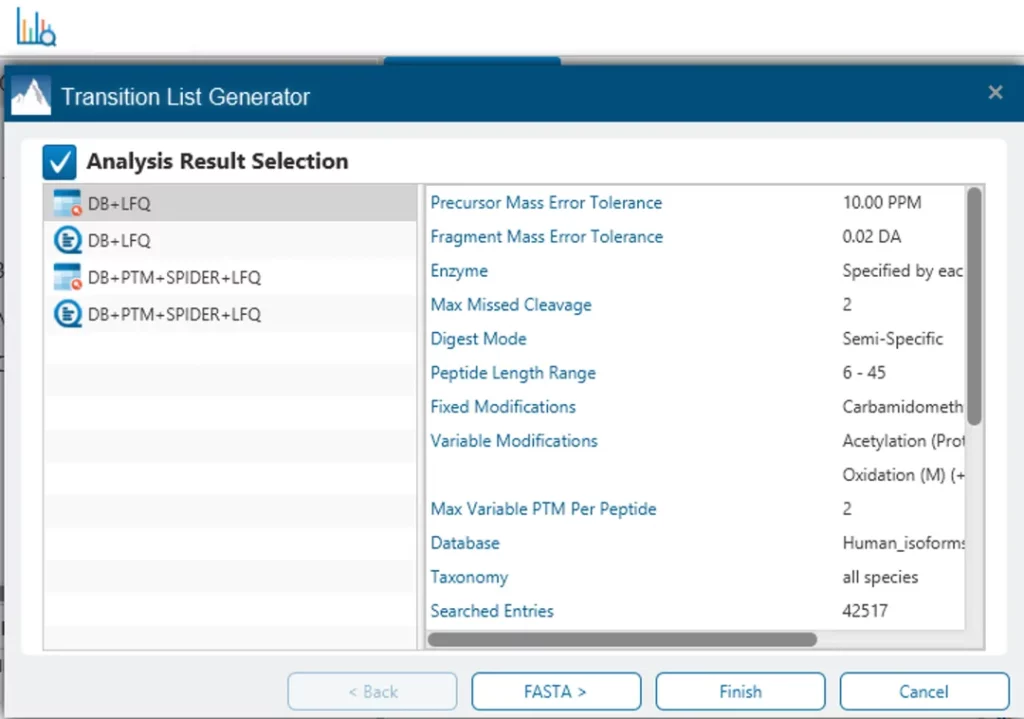
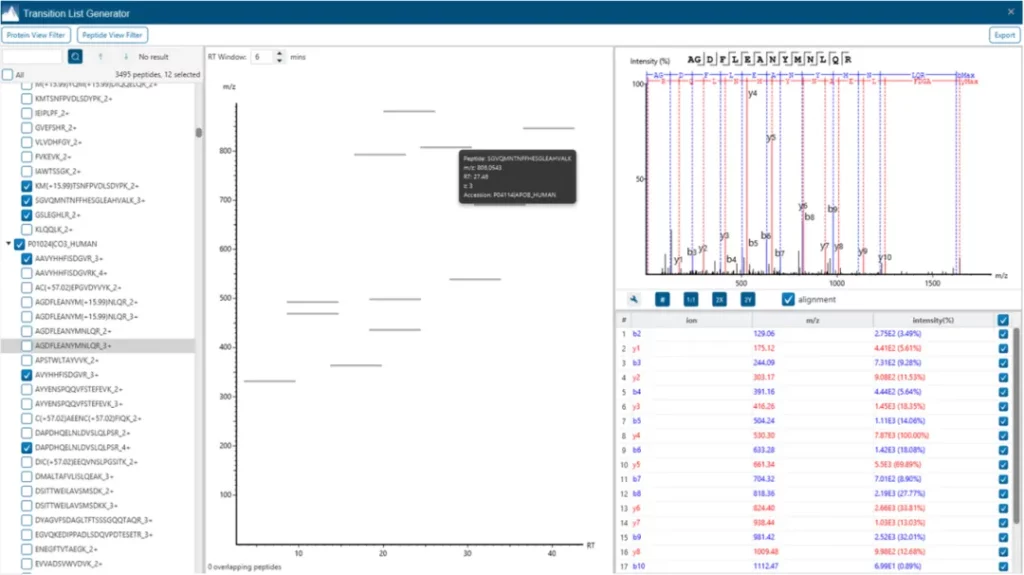
Transition list自动生成工具
>Tools列表中新增的“Transition List Generator”工具,可以直接从PEAKS分析结果或基于fasta理论酶切生成用于靶向蛋白组分析的transition列表。
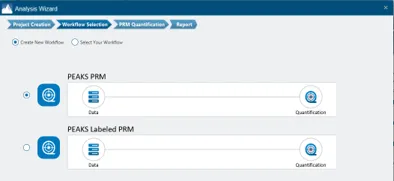
PRM定量分析
>新版本的PRM分析支持SILAC标记定量,以提升靶向定量的准确性
>新增timsTOF PRM定量分析
>PRM标准曲线定量数据变为非必选
>新增PRM蛋白注释,可展示protein匹配结果
PEAKS: 先进的蛋白质组学解决方案
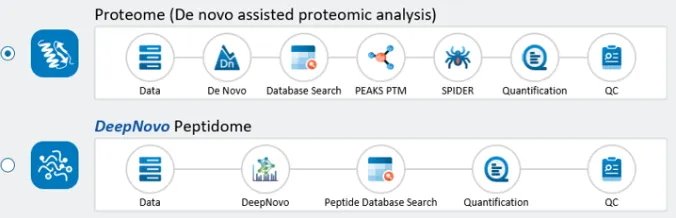
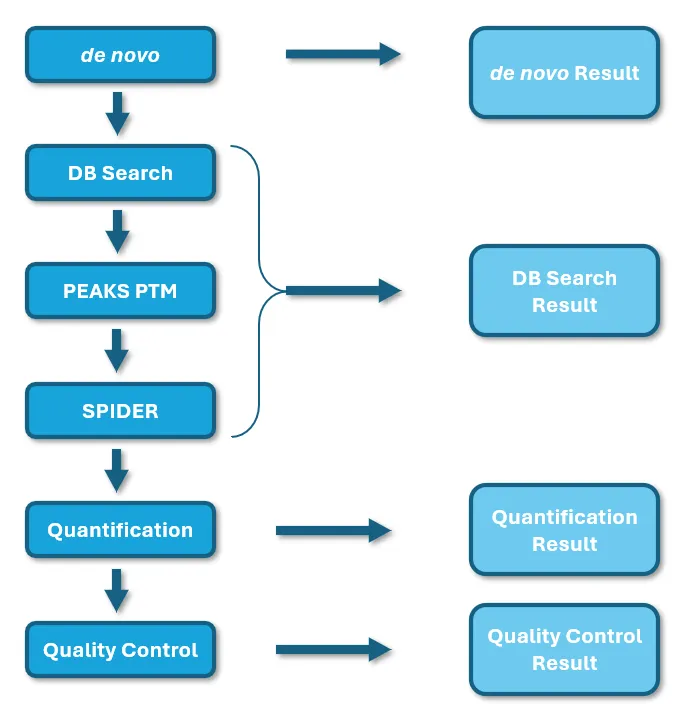
全新设计的DDA和DIA蛋白质组学和多肽组学工作流
将共有的分析步骤整个到一个工作流中,用户可根据分析需求在任意步骤选择提交分析,并可在此分析基础上添加其他步骤。
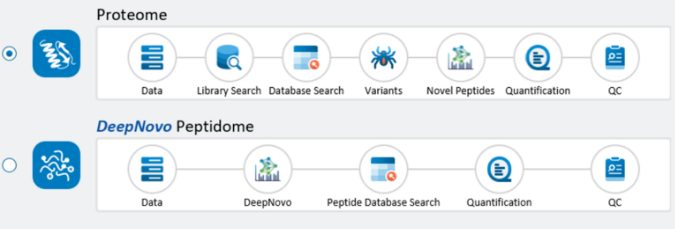
DIA序列变异分析实现更深层次的蛋白质组学发现
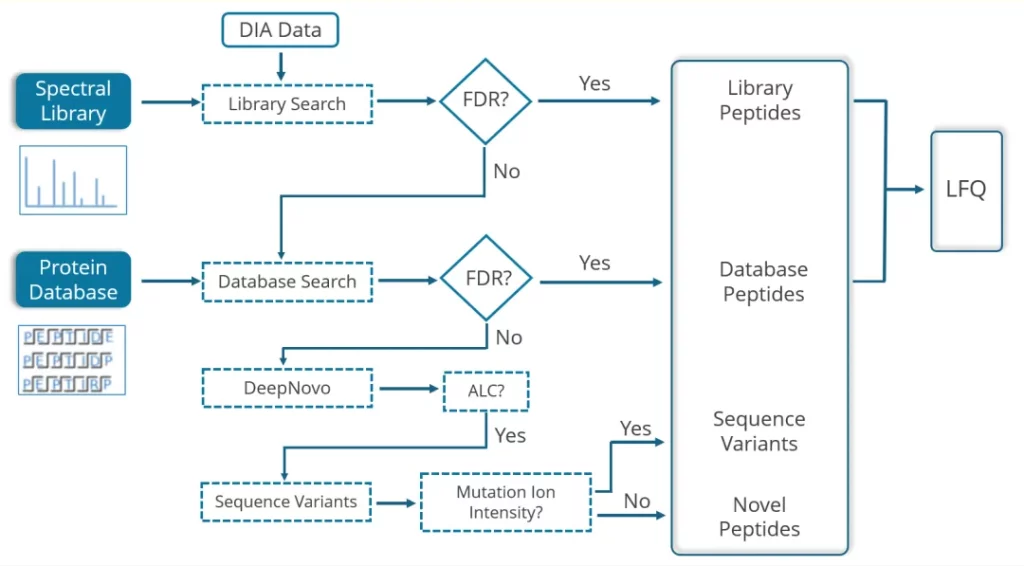
在使用小鼠数据库搜索人源的数据集的模拟测试中,PEAKS® 13 DIA可以准确的鉴定出两个物种间的突变序列和差异肽段,从而实现human肽段的正确识别[1]。
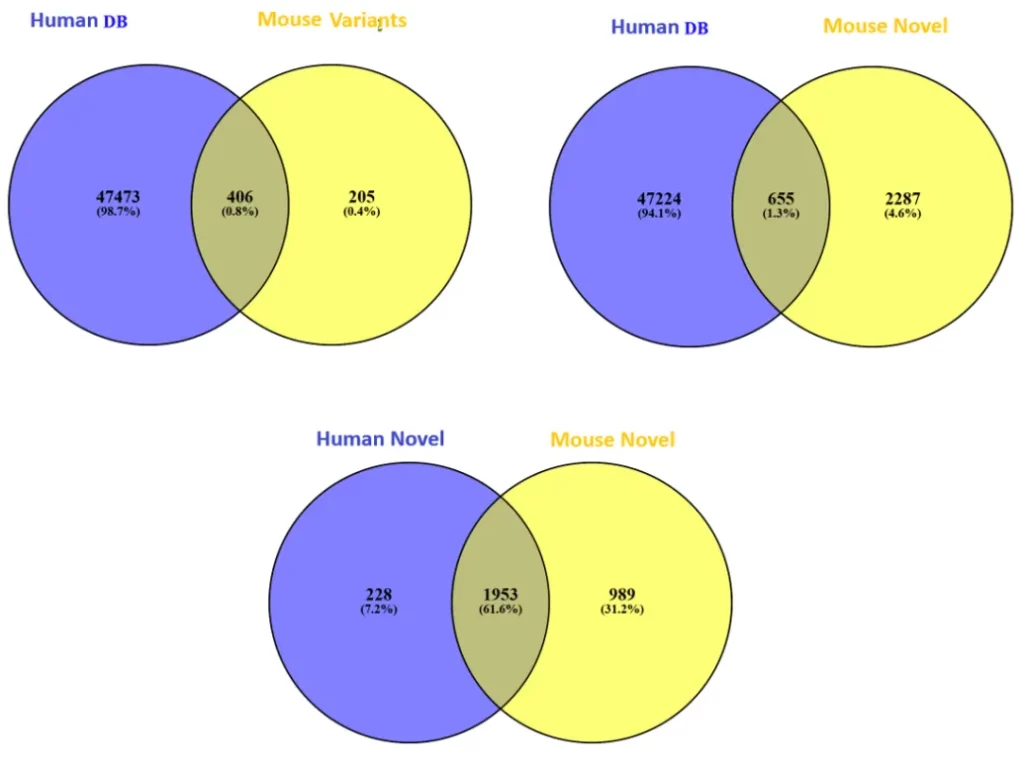
此外,我们最新的研究成果[2]展示了我们的DIA解决方案在跨物种交叉检索测试中的高度准确性,可以弥补现有常见的分析方法中的缺失。
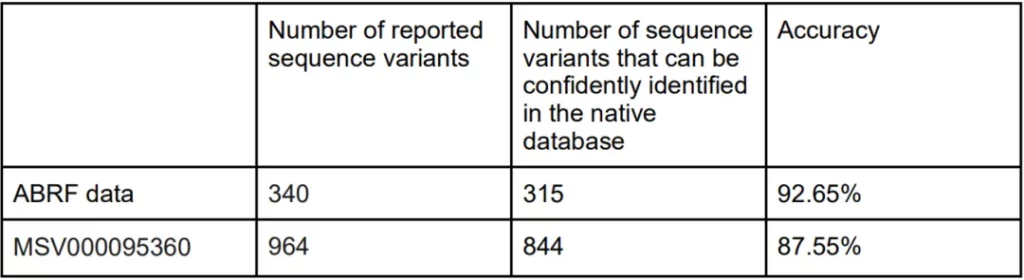
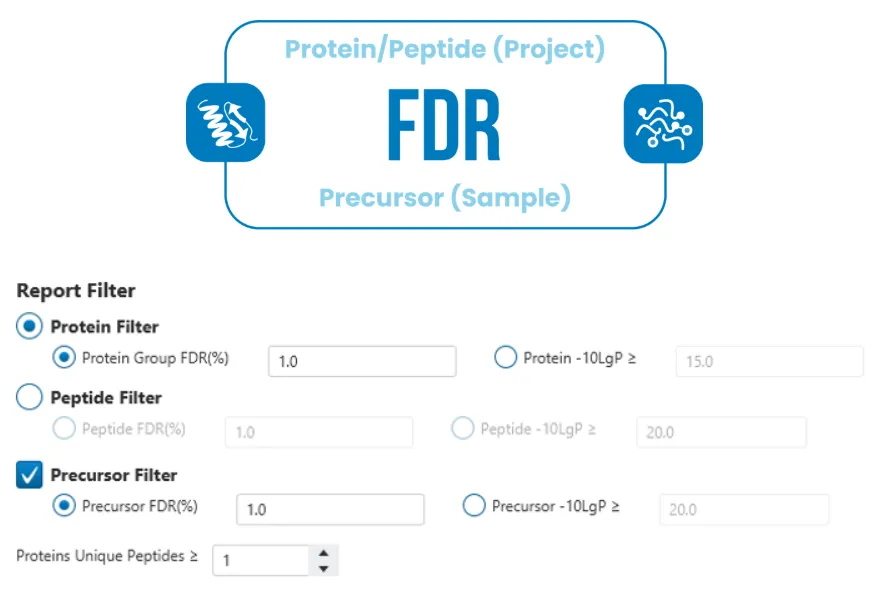
优化FDR和结果过滤方法
DIA结果过滤参数可选择基于整个project,也可基于单个sample,以适应不同的样本情况,获取最佳的鉴定结果。
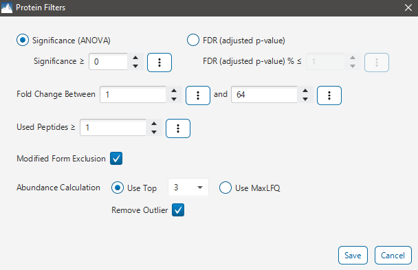
DIA蛋白质定量支持Top 3或MaxLFQ
用户可根据研究需求和数据特点选择合适的定量方法。
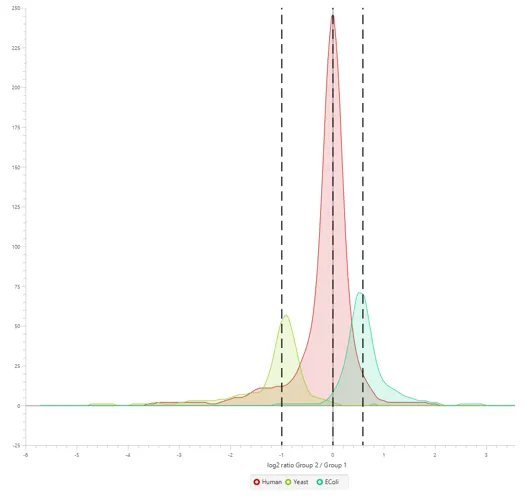
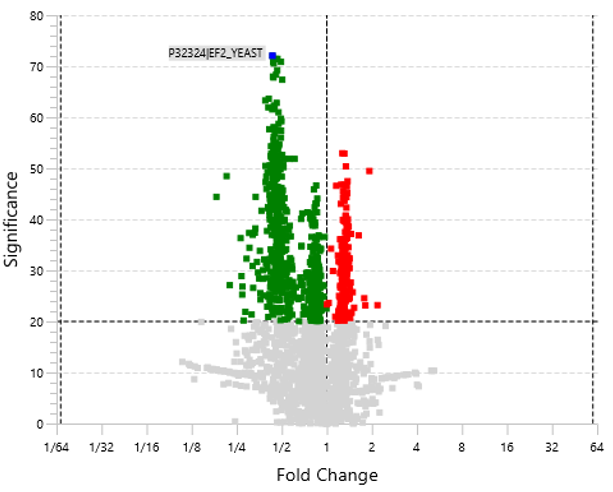
PEAKS DIA多肽组学,支持突变肽发现
最新的DIA Peptidome工作流整合了基于深度学习的GraphNovo算法进行多肽测序和序列变异分析,进一步提高定性的可信度[3]。LFQ可对数据库匹配和序列变异肽同时定量。
DDA蛋白质组学工作流
支持de novo多肽测序、数据库搜索、PEAKS PTM和SPIDER定性分析,标记和非标记定量新增对PEAKS PTM和SPIDER发现多肽的支持。每个分析步骤均有对应的可视化结果展示。
更加灵活的PRM靶向分析工作流
新版本优化了PEAKS PRM工作流,参数设置更灵活。并且,针对不同的应用场景,拆分了标记和非标记两个流程。PEAKS®13也新增了PRM PASEF(Bruker)的数据支持。
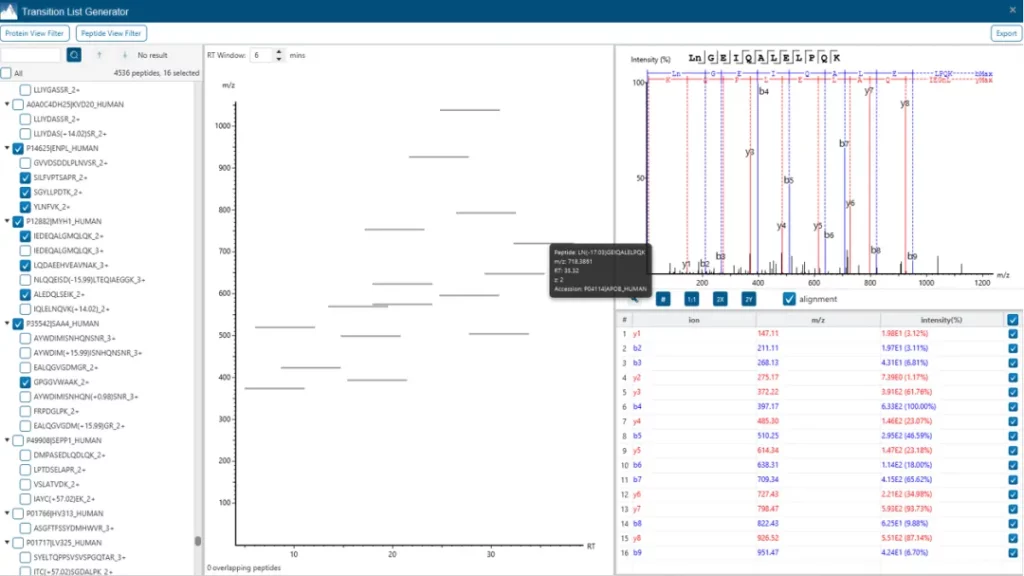
Transition list生成工具使靶向肽段选择更加便捷
新增的Transition List Generator工具可轻松实现发现蛋白组到靶向分析间的过渡,用户可以从PEAKS已有的分析结果或者参考fasta序列直接可视化碎片离子并进行Transition的选择和导出。
参考文献
- Shan, B., Korkola, N. Uncover the hidden peptidome/proteome with sequence variants and novel peptides from DIA data. Bioinformatics Solutions Inc., Waterloo, Canada
- Qiao, R., Li, H., Bian, H., Xin, L., Shan, B. De Novo sequencing-assisted homology search for DIA data analysis enables low abundance peptide variants discovery. bioRxiv (2025). doi:10.1101/2025.05.30.6570543.
- Mao, Z., Zhang, R., Xin, L. et al. Mitigating the missing-fragmentation problem in de novo peptide sequencing with a two-stage graph-based deep learning model. Nat Mach Intell 5, 1250–1260 (2023). doi:10.1038/s42256-023-00738-x